
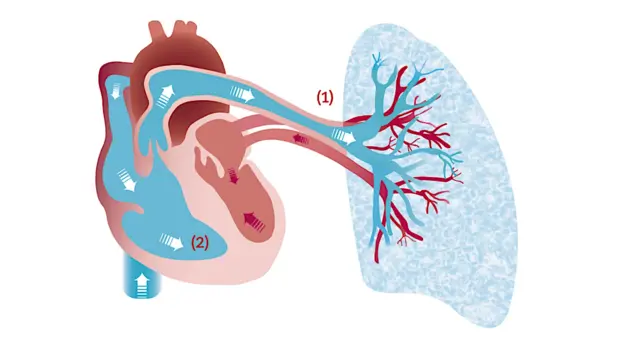
Pulmonale Hypertonie (PH) ist eine schwerwiegende Krankheit, bei der die Lunge und das Herz betroffen sind
Die Blutgefäße der Lunge sind verengt, wodurch der Blutdruck in den Lungengefäßen zwischen rechter und linker Herzkammer ansteigt. Dies führt zu einer Durchblutungsstörung der Lunge, zu einer verschlechterten Sauerstoffaufnahme und zu einer zunehmenden Überlastung der rechten Herzkammer bis hin zum Herzversagen.
Die Lebenserwartung mit pulmonaler Hypertonie nach Diagnose beträgt unbehandelt 2,8 Jahre, nach einer Studie aus den USA. Selbst manche Diagnoseuntersuchung wie der Lungenfunktionstest erlaubt keine Beurteilung des Schweregrades der Krankheit. Auch der 6-Minuten- Gehtest ist kein Maßstab für die körperliche Leistungsfähigkeit. Dieser Test wird unter „Laborbedingungen“ durchgeführt, unter Bereitstellung von klinischen Notfallmaßnahmen. Seine Ergebnisse entsprechen nicht den Alltagsleistungen. Vielmehr liegt eine erhebliche Gefahr zur Kollapsneigung beim zügigen Gehen oder Gehen von leichten Steigungen vor. Nach außen ist die Krankheit oft nicht sichtbar, was zu erheblichen Missverständnissen führen kann.
Menschen mit dieser Krankheit sind chronisch kurzatmig und körperlich wenig belastbar. Die Krankheit ist meist fortschreitend und kann unbehandelt zu einem frühzeitigen Tod führen. Viele Betroffene und Ärzte stehen der Krankheit völlig hilflos gegenüber. Bis vor wenigen Jahren war die Transplantation der Lunge oder von Herz und Lunge der einzige Ausweg. Heute hat diese Therapieoption einen anderen Stellenwert.
Oft werden die ersten Anzeichen der Krankheit, Luftnot bei Belastung und Müdigkeit, vom Betroffenen selbst nicht wahrgenommen, weil die Krankheit ganz schleichend beginnt. Ebenso ist die ärztliche Untersuchung häufig nicht richtungweisend, und es werden Verlegenheits-Diagnosen wie „mangelnder Trainingszustand“ oder „psychovegetative Erschöpfung“ gestellt.
Die Ursachen der Krankheit sind noch nicht gänzlich aufgeschlüsselt. Sie kann erblich auftreten und mehrere Generationen einer Familie betreffen. Deshalb muss den direkten Verwandten der Patienten besondere Aufmerksamkeit gelten. Neben dieser selteneren „primären“ Form des Lungenhochdrucks (früher als PPH bezeichnet, heute sog. idiopathische pulmonale arterielle Hypertonie (iPAH)) tritt Lungenhochdruck häufig in Folge chronischer Lungenerkrankungen (z. B. chronische Raucherbronchitis (COPD), Lungenfibrose), bei Bindegewebserkrankungen (CREST, Sklerodermie), Lebererkrankungen, bei der HIV Infektion oder als Folge von Lungenembolien (sog. chronisch thromboembolische pulmonale Hypertonie (CTEPH)) auf.
In Deutschland stehen bisher wenige medikamentöse Therapieoptionen zur Verfügung. Sie werden als Inhalation, als subkutane Infusion oder zur oralen Einnahme in Tablettenform angeboten. Der Kombinationstherapie kommt zunehmende Bedeutung zu. Die bisherigen Therapien wirken symptomatisch und können die Krankheit noch nicht heilen.
Die Entwicklung weiterer Therapieoptionen mit dem Ziel der Heilung bei Lungenhochdruck ist dringend geboten. Die Behandlung der Patienten sollte ausschließlich an PH spezialisierten Zentren erfolgen, da nur hier die Erfahrungen vorliegen, um Patienten alle nötigen Therapieoptionen zur Verfügung zu stellen und ihnen gegebenenfalls eine Teilnahme an klinischen Studien zu ermöglichen.
Wissenswertes
Die pulmonale Hypertonie (PH) ist eine schwerwiegende Erkrankung, die aufgrund einer starken Verengung der präkapillären Lungengefäße zu einer fortschreitenden Belastung der rechten Herzkammer führt und für die es viele verschiedene Ursachen gibt. Einige dieser Krankheiten werden als pulmonal arterielle Hypertonie (PAH) zusammengefasst. Die Ursache der idiopathischen PH (früher „primäre pulmonale Hypertonie-PPH“) ist definitionsgemäß unbekannt. Zur PH gehören auch durch Appetitzügler, Lebererkrankungen, Bindegewebserkrankungen, HIV-Infektion und angeborene Herzfehler ausgelöste Erkrankungen.
Linksherzerkrankungen, chronisch obstruktiven Bronchitis (COPD), chronisch-thromboembolischen Erkrankungen und anderen Lungenerkrankungen (z. B. Lungenfibrose), die mit einem Sauerstoffmangel einhergehen, sowie einigen sehr seltenen anderen Erkrankungen.
Frauen sind etwa doppelt so häufig betroffen wie Männer. Jedes Lebensalter ist möglich, am häufigsten ist der Krankheitsbeginn aber zwischen dem 20. und 40. Lebensjahr.
Die idiopathische PH kann vererbt werden. Bei anderen Formen der PH wird eine genetische Disposition angenommen. Immunologische Besonderheiten sind häufig nachweisbar, ihre medizinische Bedeutung ist aber unklar. Appetitzügler erhöhen das Risiko einer PH-Entstehung etwa 10-fach.
• Luftnot bei Belastung
• Vorzeitige Erschöpfung/Ermüdung bei Belastung
• Brustenge oder -schmerzen bei Belastung
• Kollaps
• Beinödeme
• Blaue Lippen